I am a passionate bioinformatician and data analyst with 9 years of working experience. I analyzed cross-sectional and longitudinal phenotypic and omic data. I also built software tools for bioinformaticians using Python and R. Sometimes, I performed wet-lab experiments.

Sep 2022 - Present
Valencia, Spain
The I2SysBio is a research institute of the Spanish National Research Institute (CSIC) focusing on basic and systems biology.
Sep 2024 - Present
Sep 2022 - Sep 2024

Feb 2019 - Nov 2022
Cologne, Germany
The MPI-AGE is one of the world leading research institutes on aging focusing on understanding the natural aging process and prevent age-related diseases.
Oct 2022 - Nov 2022
Feb 2019 - Oct 2022

Jun 2017 - Dec 2018
Valencia, Spain
Research foundation of the Clinical University Hospital, performs translational research and clinical diagnosis.
Jun 2017 - Dec 2018

Jan 2017 - Jun 2017
Valencia, Spain
Spanish leading company in the field of medical genetics.
Jan 2017 - Jun 2017
 01/02/2019-21/10/2022 Ph.D in Natural SciencesThesis title:The effect of Dietary Restriction on the Microbiome and the Adaptive Immune System in Mouse Ageing Grade:Outstanding Supervisors:Prof. Dame Linda Partridge and Prof. Andreas Beyer url:Location:Max Planck Institute for Biology of Ageing, Department of Biological Mechanisms of Aging | ||
 01/09/2016-01/10/2018 M.Sc. in BioinformaticsThesis title:Study of Genetic Alterations in Patients with Imprinting Diseases Grade:Outstanding Supervisors:Dr. Javier Chaves and Dr. Guiomar Pérez de Nanclares Location:INCLIVA, Genomics and Genetic Diagnosis Unit | ||
01/09/2015-01/07/2016 Postgraduate Diploma in Medical GeneticsLocation:Príncipe Felipe Research Center, Computational Genomics Department | ||
01/09/2011-01/07/2016 B.Sc. in BiologyThesis title:Characterization of CNVs in the Valencian Population, identified using Whole Genome Arrays Grade:Excellent Supervisor:Dr. Raquel Rodríguez-López Location:General University Hospital of Valencia, Genetics Diagnosis Unit |

Grant for developing software for differentiating between technical noise and transcript divergency in long-reads transcriptomics.

Grant for a Oxford Nanopore PromethION P2 Solo pack including sequencer and reagents. Investigating RNA degradation patterns using Nanopore long-read sequencing.


Grant for deciphering the role of RNA biological noise in aging using long-reads transcriptomics.


Development of bulk and single-cell multi-omics simulation software.

Bioinformatic pipeline development and statistical analysis for cross-sectional and longitudinal omics data.

Bioinformatic pipeline development for genomic variant prioritization.
Accounting support to Petrés Town Hall.
Accounting support to Petrés Town Hall.
Suppression of the insulin–IGF–mTORC1–Ras network ameliorates aging in animals. Many drugs have targets in the network because of its roles in cancer and metabolic disease and are candidates for repurposing as geroprotectors. Rapamycin, an established geroprotective drug, blocks mTORC1 signaling, and trametinib inhibits the Ras–MEK–ERK pathway. In this study, we assessed survival and health of male and female mice treated with trametinib, rapamycin or their combination. We show here that trametinib treatment extended lifespan in both sexes and that its combination with rapamycin was additive. Combination treatment reduced liver tumors in both sexes and spleen tumors in male mice, blocked the age-related increase in brain glucose uptake and strongly reduced inflammation in brain, kidney, spleen and muscle and circulating levels of pro-inflammatory cytokines. We conclude that trametinib is a geroprotector in mice and that its combination with rapamycin is more effective than either drug alone, making the combination a candidate for repurposing as a gerotherapy in humans.
Transcriptome sequencing revolutionized the analysis of gene expression, providing an unbiased approach to gene detection and quantification that enabled the discovery of novel isoforms, alternative splicing events and fusion transcripts. However, although short-read sequencing technologies have surpassed the limited dynamic range of previous technologies such as microarrays, they have limitations, for example, in resolving full-length transcripts and complex isoforms. Over the past 5 years, long-read sequencing technologies have matured considerably, with improvements in instrumentation and analytical methods, enabling their application to RNA sequencing (RNA-seq). Benchmarking studies are beginning to identify the strengths and limitations of long-read RNA-seq, although there remains a need for comprehensive resources to guide newcomers through the intricacies of this approach. In this Review, we provide a comprehensive overview of the long-read RNA-seq workflow, from library preparation and sequencing challenges to core data processing, downstream analyses and emerging developments. We present an extensive inventory of experimental and analytical methods and discuss current challenges and prospects.
As multi-omics sequencing technologies advance, the need for simulation tools capable of generating realistic and diverse (bulk and single-cell) multi-omics datasets for method testing and benchmarking becomes increasingly important. We present MOSim, an R package that simulates both bulk (via mosim function) and single-cell (via sc_mosim function) multi-omics data. The mosim function generates bulk transcriptomics data (RNA-seq) and additional regulatory omics layers (ATAC-seq, miRNA-seq, ChIP-seq, Methyl-seq, and transcription factors), while sc_mosim simulates single-cell transcriptomics data (scRNA-seq) with scATAC-seq and transcription factors as regulatory layers. The tool supports various experimental designs, including simulation of gene co-expression patterns, biological replicates, and differential expression between conditions. MOSim enables users to generate quantification matrices for each simulated omics data type, capturing the heterogeneity and complexity of bulk and single-cell multi-omics datasets. Furthermore, MOSim provides differentially abundant features within each omics layer and elucidates the active regulatory relationships between regulatory omics and gene expression data at both bulk and single-cell levels. By leveraging MOSim, researchers will be able to generate realistic and customizable bulk and single-cell multi-omics datasets to benchmark and validate analytical methods specifically designed for the integrative analysis of diverse regulatory omics data.
SQANTI-reads leverages SQANTI3, a tool for the analysis of the quality of transcript models, to develop a read-level quality control framework for replicated long-read RNA-seq experiments. The number and distribution of reads, as well as the number and distribution of unique junction chains (transcript splicing patterns), in SQANTI3 structural categories are informative of raw data quality. Multisample visualizations of QC metrics are presented by experimental design factors to identify outliers. We introduce new metrics for (1) the identification of potentially under-annotated genes and putative novel transcripts and for (2) quantifying variation in junction donors and acceptors. We applied SQANTI-reads to two different data sets, a Drosophila developmental experiment and a multiplatform data set from the LRGASP project and demonstrate that the tool effectively reveals the impact of read coverage on data quality, and readily identifies strong and weak splicing sites.
Long-read sequencing (LRS) technologies have revolutionized transcriptomic research by enabling the comprehensive sequencing of full-length transcripts. Using these technologies, researchers have reported tens of thousands of novel transcripts, even in well-annotated genomes, while developing new algorithms and experimental approaches to handle the noisy data. The Long-read RNA-seq Genome Annotation Assessment Project community effort benchmarked LRS methods in transcriptomics and validated many novel, lowly expressed, often times sample-specific transcripts identified by long reads. These molecules represent deviations of the major transcriptional program that were overlooked by short-read sequencing methods but are now captured by the full-length, single-molecule approach. This Perspective discusses the challenges and opportunities associated with LRS’ capacity to unravel this fraction of the transcriptome, in terms of both transcriptome biology and genome annotation. For transcriptome biology, we need to develop novel experimental and computational methods to effectively differentiate technology errors from rare but real molecules. For genome annotation, we must agree on the strategy to capture molecular variability while still defining reference annotations that are useful for the genomics community.
Rapamycin, a powerful geroprotective drug, can have detrimental effects when administered chronically. We determined whether intermittent treatment of mice can reduce negative effects while maintaining benefits of chronic treatment. From 6 months of age, male and female C3B6F1 hybrid mice were either continuously fed with 42 mg/kg rapamycin, or intermittently fed by alternating weekly feeding of 42 mg/kg rapamycin food with weekly control feeding. Survival of these mice compared to control animals was measured. Furthermore, longitudinal phenotyping including metabolic (body composition, GTT, ITT, indirect calorimetry) and fitness phenotypes (treadmil, rotarod, electrocardiography and open field) was performed. Organ specific pathology was assessed at 24 months of age. Chronic rapamycin treatment induced glucose intolerance, which was partially ameliorated by intermittent treatment. Chronic and intermittent rapamycin treatments increased lifespan equally in males, while in females chronic treatment resulted in slightly higher survival. The two treatments had equivalent effects on testicular degeneration, heart fibrosis and liver lipidosis. In males, the two treatment regimes led to a similar increase in motor coordination, heart rate and Q-T interval, and reduction in spleen weight, while in females, they equally reduced BAT inflammation and spleen weight and maintained heart rate and Q-T interval. However, other health parameters, including age related pathologies, were better prevented by continuous treatment. Intermittent rapamycin treatment is effective in prolonging lifespan and reduces some side-effects of chronic treatment, but chronic treatment is more beneficial to healthspan.
Aging impairs the capacity to respond to novel antigens, reducing immune protection against pathogens and vaccine efficacy. Dietary restriction (DR) extends life- and health span in diverse animals. However, little is known about the capacity of DR to combat the decline in immune function. Here, we study the changes in B cell receptor (BCR) repertoire during aging in DR and control mice. By sequencing the variable region of the BCR heavy chain in the spleen, we show that DR preserves diversity and attenuates the increase in clonal expansions throughout aging. Remarkably, mice starting DR in mid-life have repertoire diversity and clonal expansion rates indistinguishable from chronic DR mice. In contrast, in the intestine, these traits are unaffected by either age or DR. Reduced within-individual B cell repertoire diversity and increased clonal expansions are correlated with higher morbidity, suggesting a potential contribution of B cell repertoire dynamics to health during aging.
Torque teno virus, the major member of the genus Alphatorquevirus , is an emerging biomarker of the net state of immunosuppression after kidney transplantation. Genetic diversity constitutes a main feature of the Anelloviridae family, although its posttransplant dynamics and clinical correlates are largely unknown. The relative abundance of Alphatorquevirus , Betatorquevirus , and Gammatorquevirus genera was investigated by high-throughput sequencing in plasma specimens obtained at various points during the first posttransplant year (n = 91 recipients). Total loads of all members of the Anelloviridae family were also quantified by an “in-house” polymerase chain reaction assay targeting conserved DNA sequences (n = 195 recipients). In addition to viral kinetics, clinical study outcomes included serious infection, immunosuppression-related adverse event (opportunistic infection and cancer)’ and acute rejection. Alphatorquevirus DNA was detected in all patients at every point, with an increase from pretransplantation to month 1. A variable proportion of recipients had detectable Betatorquevirus and Gammatorquevirus at lower frequencies. At least 1 change in the predominant genus (mainly as early transition to Alphatorquevirus predominance) was shown in 35.6% of evaluable patients. Total anelloviruses DNA levels increased from baseline to month 1, to peak by month 3 and decrease thereafter, and were higher in patients treated with T-cell depleting agents. There was a significant albeit weak-to-moderate correlation between total anelloviruses and TTV DNA levels. No associations were found between the predominant Anelloviridae genus or total anelloviruses DNA levels and clinical outcomes. Our study provides novel insight into the evolution of the anellome after kidney transplantation.
Genome assembly of viruses with high mutation rates, such as Norovirus and other RNA viruses, or from metagenome samples, poses a challenge for the scientific community due to the coexistence of several viral quasispecies and strains. Furthermore, there is no standard method for obtaining whole-genome sequences in non-related patients. After polyA RNA isolation and sequencing in eight patients with acute gastroenteritis, we evaluated two de Bruijn graph assemblers (SPAdes and MEGAHIT), combined with four different and common pre-assembly strategies, and compared those yielding whole genome Norovirus contigs. Reference-genome guided strategies with both host and target virus did not present any advantages compared to the assembly of non-filtered data in the case of SPAdes, and in the case of MEGAHIT, only host genome filtering presented improvements. MEGAHIT performed better than SPAdes in most samples, reaching complete genome sequences in most of them for all the strategies employed. Read binning with CD-HIT improved assembly when paired with different analysis strategies, and more notably in the case of SPAdes. Not all metagenome assemblies are equal and the choice in the workflow depends on the species studied and the prior steps to analysis. We may need different approaches even for samples treated equally due to the presence of high intra host variability. We tested and compared different workflows for the accurate assembly of Norovirus genomes and established their assembly capacities for this purpose.
Monitoring of alphatorquevirus (torque teno virus [TTV]) DNA in plasma may prove to be useful to assess the net state of immune competence following allogeneic hematopoietic stem cell transplantation (allo-HSCT). There are scarce data published on the prevalence of beta (torque teno mini virus [TTMV]) and gammatorqueviruses (torque teno midi virus [TTMDV]) and, in particular, on the dynamics of anelloviruses in allo-HSCT patients. Twenty-five allo-HSCT recipients with available plasma specimens obtained before conditioning and after engraftment were included. Degenerated primers targeting a highly conserved genomic sequence across all anelloviruses were designed for genomic amplification and high-throughput sequencing. Co-detection of TTV, TTMV, and TTMDV both in pre-transplant and post-engraftment plasma specimens was documented in more than two-thirds of patients. The use of quantitative real-time polymerase chain reaction (PCR) assays targeting TTMV and TTMDV in addition to TTV may add value to TTV-specific PCR assays in the inference of the net state of immunosuppresion or immune competence in this clinical setting.
Jacobsen syndrome or JBS (OMIM 147791) is a contiguous gene syndrome caused by a deletion affecting the terminal q region of chromosome 11. The phenotype of patients with JBS is a specific syndromic phenotype predominately associated with hematological alterations. Complete and partial JBS are differentiated depending on which functional and causal genes are haploinsufficient in the patient. We describe the case of a 6-year-old Bulgarian boy in which it was possible to identify all of the major signs and symptoms listed by the Online Mendelian Inheritance in Man (OMIM) catalog using the Human Phenotype Ontology (HPO). Extensive blood and marrow tests revealed the existence of thrombocytopenia and leucopenia, specifically due to low levels of T and B cells and low levels of IgM. Genetic analysis using whole-genome single nucleotide polymorphisms (SNPs)/copy number variations (CNVs) microarray hybridization confirmed that the patient had the deletion arr[hg19]11q24.3q25(128,137,532–134,938,470)x1 in heterozygosis. This alteration was considered causal of partial JBS because the essential BSX and NRGN genes were not included, though 30 of the 96 HPO identifiers associated with this OMIM were identified in the patient. The deletion of the FLI-1, ETS1, JAM3 and THYN1 genes was considered to be directly associated with the immunodeficiency exhibited by the patient. Although immunodeficiency is widely accepted as a major sign of JBS, only constipation, bone marrow hypocellularity and recurrent respiratory infections have been included in the HPO as terms used to refer to the immunological defects in JBS. Exhaustive functional analysis and individual monitoring are required and should be mandatory for these patients.
Human noroviruses are responsible for most nonbacterial acute gastroenteritis cases. The GII.2, GII.4, and GII.17 genotypes of human noroviruses have recently arisen as the most frequent genotypes found in humans worldwide. We report here seven nearly complete genomes of these genotypes from patients with acute gastroenteritis in Valencia, Spain.
Since genomic SNPs/CNVs arrays were implemented as a diagnostic tool in clinical settings to search for the cause of idiopathic intellectual disability, chromosomal imbalances have been precisely described as being the cause of many new syndromes, especially when they are associated with multiple congenital anomalies and/or dimorphism. High-density SNPs/CNVs microarray was used to delineate genotype-phenotype correlation in a 2.5 year-old girl who carried a mosaic characterized by a predominant cell line representing 92% which carried a duplication of 12.5 Mb of the 18p11.32p11.21 chromosomal region (chr18:12602631_telomeric) and a deletion and a deletion of 20.3 Mb of the 18q21.32q23 chromosomal region (chr18:57691236_telomeric) and a second minor cell line (8%) presented a ring chromosome, carrying the deletion of 20.3 Mb of the 18q21.32q23 chromosomal region. The microarray analysis identified the genetic causes for the specific phenotype of the patient, whose more evident signs and symptoms were mainly associated to the genomic regions duplicated and in haploinsufficiency. Although the conventional karyotype is currently considered a diagnostic technique of support or second line, it was key to establish the etiology of the alteration that the patient carried. Its performance allowed the correct interpretation of the result of the array, whose characterization facilitated its correlation with the phenotypic features of the patient. From both tests, it was possible to conclude the existence of ring chromosome 18, as well as the percentage of the mosaic lines. The deleterious capabilities of the affected OMIM and dominant genes were evaluated along her period of life. A rigorous clinical following up in that patient included valorous phenotypic data to the array data bases, and will make less difficult to hypothesize about prognosis in other individuals who carry overlapping similar high risk genetic alterations.
Human noroviruses are the most common cause of nonbacterial acute gastroenteritis worldwide. We report here the nearly complete genome sequence (7,551 nucleotides) of a human norovirus GII.P17-GII.17 strain detected in July 2015 in the stool sample from an adult with acute gastroenteritis in Brazil.
The first step in any genetic analysis is DNA collection and processing. Each type of genetic analysis requires specific processing, so the path from DNA to data is not always the same, making it essential to provide appropriate material for each analysis. This chapter reviews the steps from DNA collection to the raw data that will enable subsequent clinical interpretation.
Reduced genetic variability in isolated populations promotes the prevalence of long contiguous stretches of homozygosity (LCSH) that may carry deleterious mutations, manifesting recessive syndromes such as Alström syndrome (OMIM 203800), caused principally by mutations in exons 8, 10, and 16 and deletions/insertions along the ALMS1 gene. Here, Sanger sequencing of these exons and whole-genome copy-number variants/single-nucleotide polymorphisms (SNPs) microarray were used to characterize ALMS1 gene in a 19-year-old Pakistani female with Alström syndrome. Sequencing did not reveal pathogenic alterations but described a set of homozygous polymorphisms. The microarray revealed these SNPs were included in an 8.24 Mb LCSH in 2p12.2p13 that contained a homozygous deletion including exons 13-16 of ALMS1 gene. Therefore, reduced genetic variability in Pakistani population enhanced the inheritance of the homozygous deletion causing Alström syndrome. The comparison of the deletion with known deletions spanning exons 13-16, described on Middle Eastern patients, suggested a fixation of the deletion in a specific haplotype.
Hereditary hemochromatosis is a disease responsible for excess blood iron. The hemochromatosis gene has two predominant variants, H63D and C282Y single nucleotide polymorphisms. Our study aims to analyze the diagnostic utility of genotyping the 63 and 282 loci, and examine the geographic distribution of these mutations in Spain. Genotyping was performed on 94 healthy control individuals and 324 patients suspected of hereditary hemochromatosis, and also biochemical test to 313 individuals in the patients group. The comparison of allelic frequencies between East and West of Spain, as well as other countries located at a similar longitude, evidenced a west-east distribution gradient of the C282Y allele. In addition, heterogeneous distribution of the H63D mutation in Spain was observed. Patients who carried the 282YY genotype showed significantly higher biochemical parameters (ferritin>300μg/L, Fe>180μg/L, IST>60%, UIBC>355μg/L and CTFH>370μg/dL), which confirmed the correlation between the mutated homozygous genotype and the associated hemochromatosis phenotype. Our results strengthen the importance of executing genetic tests to increase the efficiency of hereditary hemochromatosis diagnosis, which reveal an interesting variability among geographical regions.
To describe the populational distribution of the UGT1A1*28 variant (genetic variant code rs8175347) located in the promotor of the UGT gene and correlate its genotypes with the results of the fasting test, as well as its relationship with the biochemical disorder of Gilbert’s syndrome (GS) in a Valencian population. We studied the prevalence of the genotypes (TA)6/6 (TA)6/7 and (TA)7/7 of the deleterious variant rs8175347 in 144 patients with hyperbilirubinemia, 38 of whom had previously undergone the fasting test to diagnose GS, and in 150 control patients. By analysing the genomic region of the TATA box of the UGT1A1 gene promotor using Sanger sequencing, we established the correlation between the rs8175347 genotypes and the fasting test results and with the patients’ biochemical disorders. The rate of heterozygosity of allele (TA)7 in the control population was 32% and increased to 87.59% among the patients with suspected GS. The rate of genotype TA7/7 was 81.94% among the patients with hyperbilirubinemia, compared with 11.33% in the control patients. The fasting test showed a 15.79% rate of false negatives and a 5.26% rate of false positives. The high frequency of allele (TA)7 among the Valencian control population, almost double the 5% reported for European control patients, confirms the high rate of GS reported in the Spanish population, without observing significant differences between the geographical ends of the country. The efficacy and reliability of the fasting test for the diagnosis of GS is questionable.
Fragile X Syndrome is the most common monogenic cause of intellectual disability. It originates by the expansion of the CGG triplet repeat in the 5’ non-coding region of FMR1 gene. We analyzed aforementioned region of susceptibility in relatives of a patient carrying an expansion in range of mutation, with the aim of identifying other carriers of deleterious alleles and offering genetic counseling. Alleles in range of premutation were found in the mother and sister (maternal half-sister) of the patient, with 106 and 60 repetitions respectively; none showed any phenotypic trait. The probability of inheriting a contracted premutated allele when maternally transmitted is low (approximately 0.76%). Therefore, given the finding of a lower number of repetitions in the offspring of a woman, the possibility of a mosaic allele product of a undetected mutated allele has to be ruled out in first stand; for this the employment of a technique of maximum sensitivity is essential. In our case it was impossible to obtain DNA samples from male parents; hence we proceeded with microsatellite analysis on the X chromosome to assign haplotypes segregating with the corresponding expanded alleles, trying to describe their heritability in the pedigree. We conclude that the premutation carried by the sister of the diagnosed patient, was inherited through her paternal route and therefore comes from a second deleterious allele, different from the one transmitted from her mother to her affected sibling. Haplotype structure identified in both carrier chromosomes, the individuals’ population of origin and the heritability by paternal branches, suggests a shared ancestor in previous generations.

Talk about MOSim at the XV Bioinformatics Symposium.

Talk about SQANTI-reads at the XV Bioinformatics Symposium.

Interview and summary about the relevance of my recent publication on long-reads.

Introductory session on study design, sequencing technologies and ground truth data.

Post by the Spanish Regional Student Group in bioinformatics about my latest published work.

Interview about the relevance of my recent publication in Nature Reviews Genetics.

Talk about the activities of the Junior Bioinformatics and Computational Biology Hub at CSIC.

Post by the Spanish Regional Student Group in bioinformatics about my latest published work.

Flash talk about my project on transcript divergency in aging using long-reads sequencing data.

Talk about the activities of the Junior Bioinformatics and Computational Biology Hub at CSIC.

Seminar about my PhD work, finalist to the “Best PhD thesis”.

Talk about women in science to senior high-school students at the Luis Amigó school in Paterna, Valencia.

Introduce myself and my research topic.

Talk about women in science to junior high-school students at the school in La Canal de Navarrés, Valencia.

Introduce myself and my research topic.

Organization of a long-reads bioinformatics workshop, from library preparation to data analysis and interpretation.

Flash talk about my published work MOSim, a bulk and single-cell multi-omics data simulator.

Lecture about the functional iso-transcriptomics pipeline (Bioinformatics).
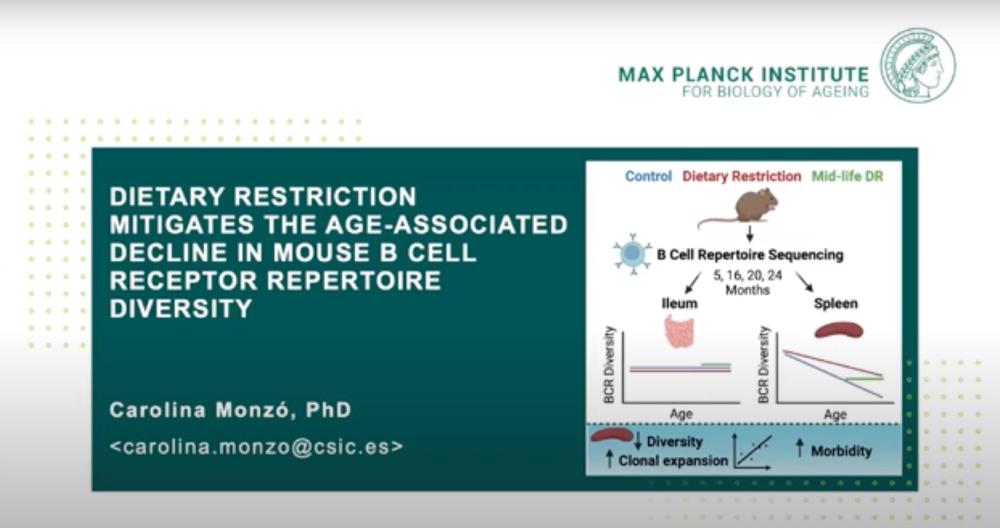
Talk about my published work on B-cells during aging and under anti-aging interventions.

Talk about my published work MOSim, a bulk and single-cell multi-omics data simulator.

Talk about my career and give tips and tricks to bioinformatics students of Valencia.
Talk about the brain drain.
Lecture introducing wet-lab aging scientists to the process of going from RNA extraction to count data (bioinformatics).
Interview about the difficulties of performing science and my latest work on fragile X syndrome.
Course on designing prompts for everyday work tasks.
Course on writing prompts for AI writing.
Core AWS concepts and the benefits of using AWS Cloud. AWS global infrastructure basics. Key AWS services (compute, networking, databases, storage). Financial aspects of AWS (cost models, billing, pricing, and tools to manage expenses)
Certification course for Function D, experimental and process design, recognized by the Spanish ministry of science.
Training course to teach researchers the foundations of good peer review.
Training in drafting and writing a fellowship proposal.
Training on identifying job offers in industry, CV and interview preparation.
Training in improving genetic dianosis of patients with rare diseases.
Training on efficient poster design and presentation.
Advanced training on drafting and writing scientific articles.
Course on machine learning methods for bioinformatics, including supervised and unsupervised methods.
Course on good scientific practices and cases discussion.
Computational methods, algorithms and data structures for analyzing DNA sequencing data.
Training to use bioinformatics to explore DNA sequences and protein functions, to evaluate virulence.
Training course on using genomics for diagnosis and cancer treatment.
Training on exploring biological functions through network analysis.
Training on using tools such as samtools, bedtools, bwa, vep etc for genomic data analysis.
Training on using python classes and packages for genomic data analysis.
Introductory training on using tools for genomic and transcriptomic data analysis.
Theoretical and practical course on identifying and classifying sharks and fish.
Theoretical and practical course on correct dissection of sharks.
Theoretical course on identification and classification of marine invertebrates, focused on cnidaria.